On 26 May 2017, the new EU Medical Devices Regulation (MDR) and In Vitro Diagnostics Regulation (IVDR) entered into force. In order to aid preparations for the provisions taking effect, the Medicines and Healthcare products Regulatory Agency (MHRA) has published materials to help manufacturers understand the new requirements, and in particular, has published an introductory Interactive Guide to the Regulations. The MHRA’s director of Medical Devices, John Wilkinson, explained that “We live in an increasingly digital world, and the way we provide our guidance is changing. We want to help manufacturers to comply with the new regulations as easily and as early as possible.”
The Interactive Guide allows users to navigate through key topics and provides a high level overview of the Regulations for manufacturers who may be looking at them for the first time, and also seeks to help experienced manufacturers navigate the changes. A brief summary of the key points is set out below.
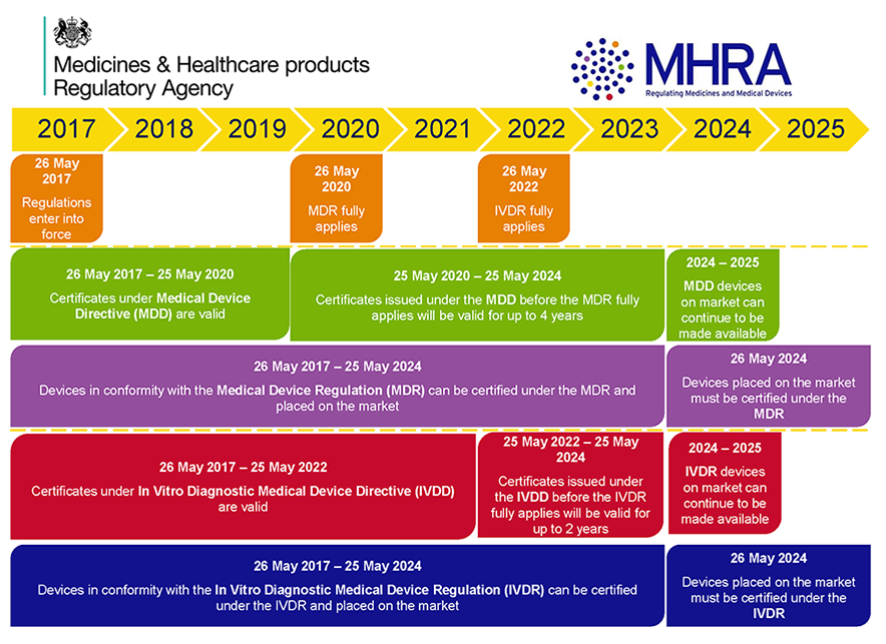
- Introduction: the majority of the MDR and IVDR provisions will apply from 26 May 2020 and 26 May 2022 respectively. The transitional provisions are complex as there are no “grandfathering” provisions for devices that are currently on the market, and all devices must comply with the new rules. Alongside the Interactive Guide, the MHRA has provided the following timeline of the transition periods:
- Definitions: as well as setting out the definitions of devices and in vitro diagnostic devices, the Interactive Guide provides guidance on borderline products and aesthetic products, such as non-corrective contact lenses and dermal fillers, which are covered by the Regulations for the first time.
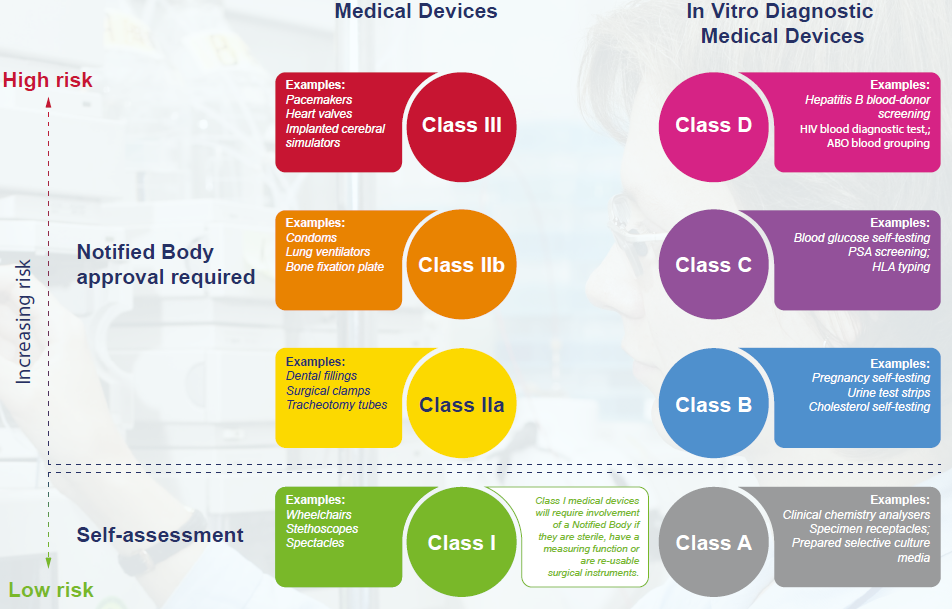
- Classification: both the MDR and the IVDR require classification based on risk, and the IVDR introduces major changes for the classification of in vitro diagnostic products in particular. The Interactive Guide provides an overview of the classification system, and includes the following useful summary:
- Conformity assessment: the Interactive Guide outlines the requirements that must be met and where they can be found in the Regulations, including the general safety and performance requirements, technical documentation and the harmonised standards/ common specifications.
- Unique Device Identifier: the Regulations include a greater emphasis on traceability throughout the whole supply chain, and in particular, introduce a unique device identification (UDI) system that will provide “a consistent and standard way to identify medical devices throughout their distribution and use by healthcare providers and patients”. Information on the UDI, manufacturers and suppliers of devices must be included on the new European databank on medical devices (Eudamed).
- Supply chain obligations: the Interactive Guide provides high-level guidance for manufacturers, authorised representatives, importers and distributors, and their new obligations under the Regulations. For example, manufacturers must have at least one person responsible for regulatory compliance, and must ensure that adequate financial coverage and stringent quality management systems are in place.
- Post-Market Surveillance and vigilance: the Regulations set out more rigorous vigilance reporting requirements, including new reporting timescales, and clearer requirements on what the post-market surveillance system should comprise of. The Interactive Guide explains that the key obligations are to ensure the ongoing safety of devices, to conduct field safety corrective actions where safety issues are identified, and to report any serious incidents or trends that are detected.